

A personalized multi-omics discovery and validation platform for recurrent head and neck squamous cell carcinoma (POLARES)
{kind=link}
Giger group Prof. Dr. med. Roland Giger
Diagnostic and therapeutic developments in recent years have improved the prognosis for patients with head and neck squamous cell carcinoma (HNSCC). Despite these developments, a significant proportion of patients relapse after an initial response to standard treatment. Salvage treatment options are limited, and personalized treatment approaches that consider the genomic/epigenetic landscape of the tumor are lacking.
The goal of this research is to establish a center of excellence in HNSCC that bridges the gap between genomic analysis and translation of findings into clinical trials. By establishing a multi-omics discovery and validation platform under the umbrella of the University Comprehensive Cancer Center Inselspital (UCI), this consortium (ORL, Head and Neck Surgery; Medical Oncology; Radiation-Oncology) aims to determine how alterations at the genomic and epigenetic level regulate carcinogenesis, treatment response and resistance in HNSCC and thereby identify novel mechanisms to target tumor relapse.
On behalf of the Consortium: Prof. Dr. Roland Giger (Lead), Otorhinolaryngology, Head and Neck Surgery; PD Dr. Olgun Eliçin, Radio-Oncology; Dr. Simon Häfliger, Medical Oncology; PD Dr. Michaela Medová, Radio-Oncology, DBMR; Prof. Dr. Carsten Riether, Medical Oncology, DBMR; Dr. Daniel H. Schanne, Radio-Oncology
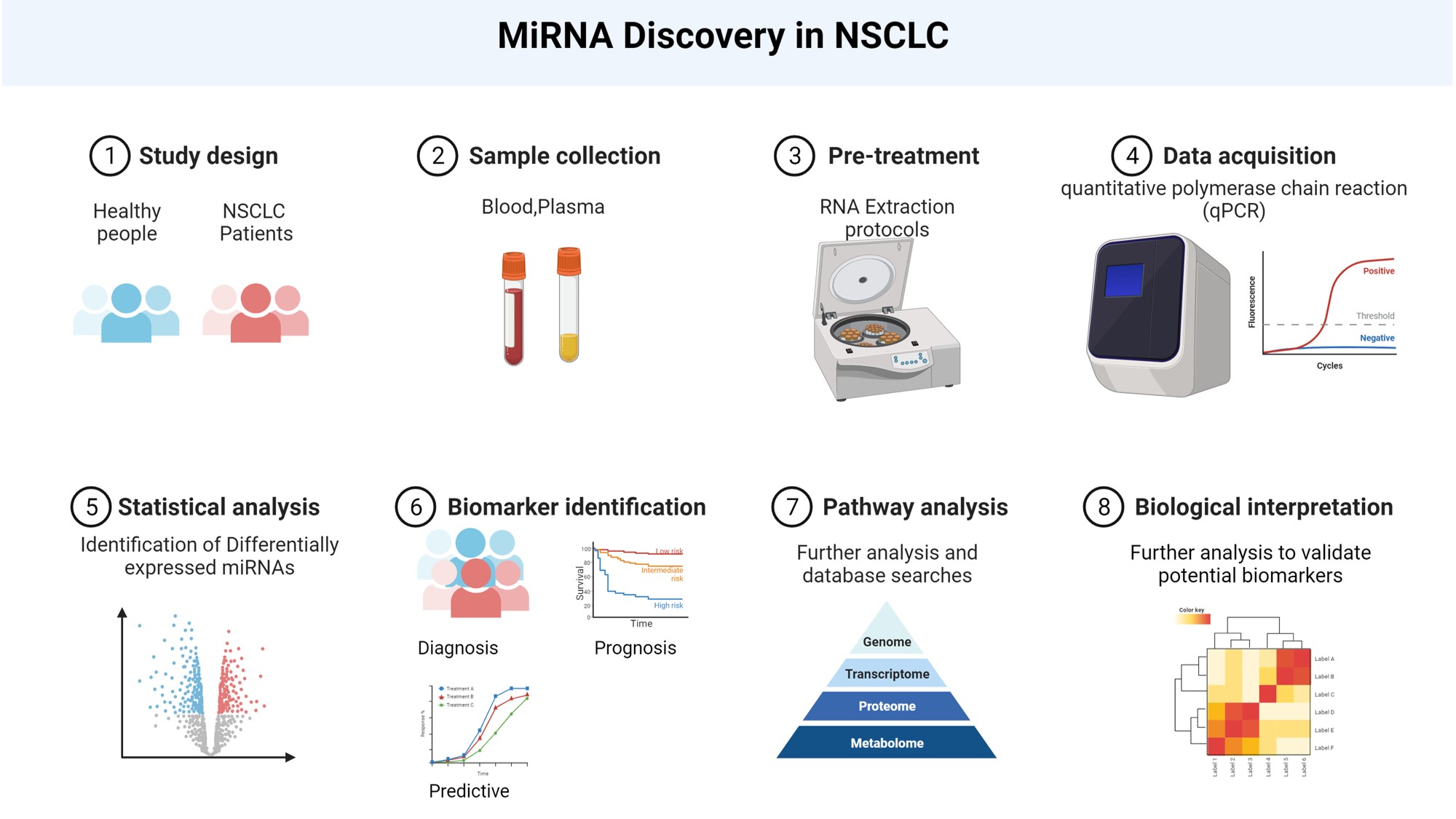
Lung Cancer and Non-Coding RNAs (ncRNAs)
{kind=link}
Häfliger group Dr. med. et phil. nat. Simon Häfliger
Lung cancer is the leading cause of cancer related death worldwide. Our research delves into the intriguing world of non-coding RNAs (ncRNAs), RNA molecules that do not translate into proteins.
- MicroRNAs (miRNAs): These tiny RNA molecules can act as either tumor suppressors or oncogenes, influencing cancer development.
- Long Non-Coding RNAs (lncRNAs): These longer RNA molecules impact gene expression and are central to epigenetic regulation.
Using public databases, blood of lung cancer patients, cancer cell lines and bioinformatics tools, we investigate specific ncRNAs with the aim to explore:
- Diagnostic Biomarkers: Can circulating ncRNAs serve as early indicators of lung cancer?
- Mechanisms of Action: How do these ncRNAs influence cancer progression?
- Predictive and Prognostic Markers: Can they guide treatment decisions?
In summary, our work bridges the gap between complex scientific research and practical applications, unraveling the mysteries of ncRNAs in lung cancer to improve patient outcomes and treatment strategies.
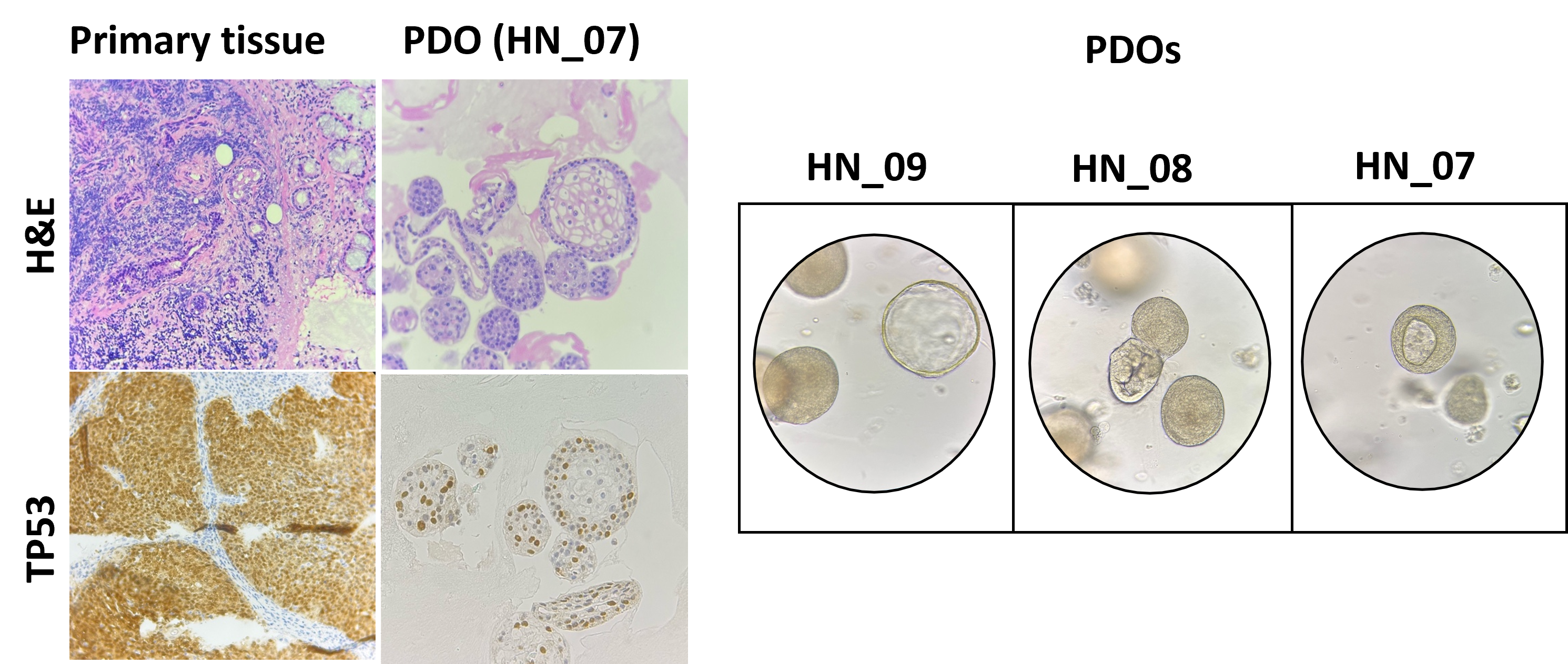
Dissecting the role of tumor cell heterogeneity in Pancreatic Neuroendocrine Tumor progression
Group Marinoni, Perren, Sadowski PD Dr. Ilaria Marinoni, PhD, Prof. Dr. med. Aurel Perren, PD Dr. Martin Sadowski, PhD
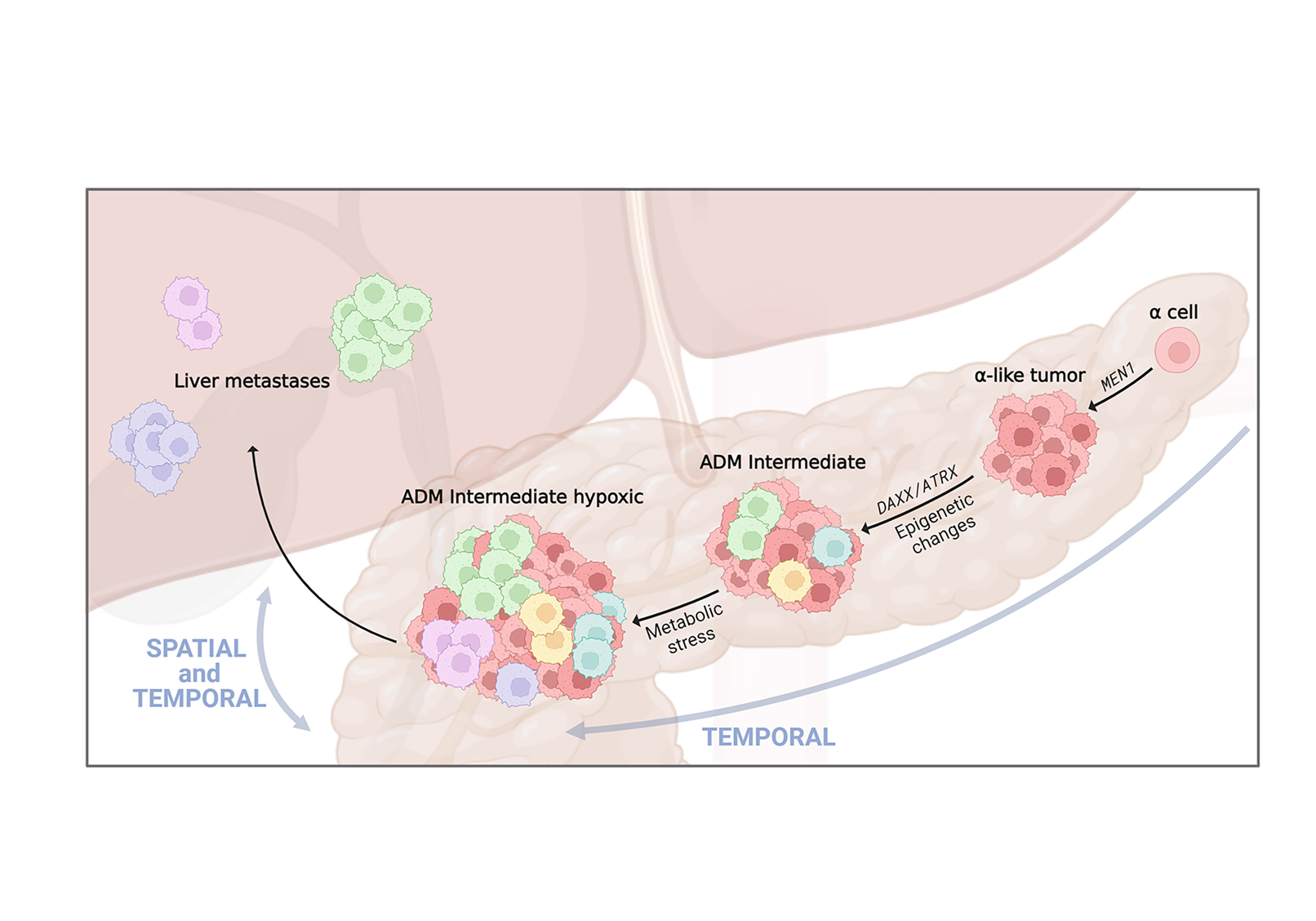
Cancer is a dynamic disease; genetic and epigenetic alterations drive intra-tumoral cell heterogeneity, resulting in the selection of aggressive cell populations capable of driving progression and ultimately metastasis.
Pancreatic neuroendocrine tumours (PanNETs) are tumours that arise from the islets of Langerhans. They exhibit intra-tumoral cell heterogeneity, but it is unclear how this evolves during tumour development and how it contributes to progression.
Our previous data suggest that epigenetic changes are the major drivers of progression and cell heterogeneity in PanNETs. By integrating epigenetic and transcriptomic profiles, we found that cell dedifferentiation and metabolic changes characterise the progression from small PanNETs to more advanced ones.
We are currently investigating the evolution of intra-tumoral heterogeneity of PanNETs through space and time. Specific cell subpopulations identified as driving progression could then be targeted to stop metastasis formation. The identification of targetable pathways that impair metastasis formation will provide a rationale for new treatments.
{kind=link}
Simple and accurate quantification of mutational signatures in tumors
Medo Group Prof. Dr. Matúš Medo
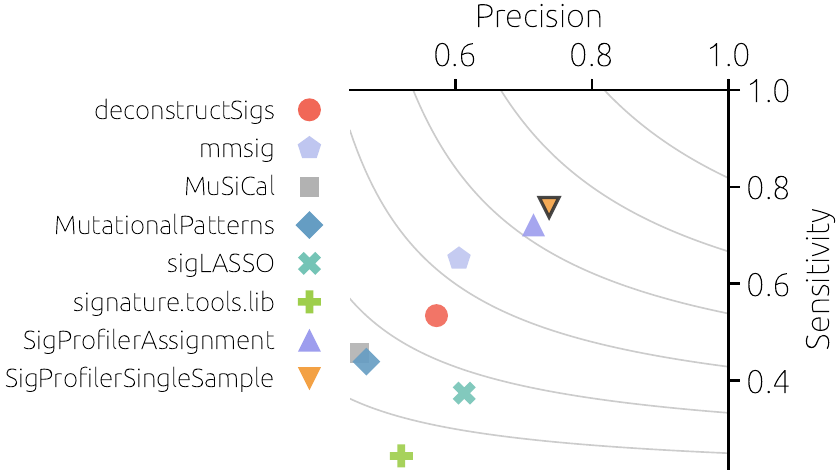
Mutations in the genome are often not random but follow distinct patterns. These patterns, termed mutational signatures, are footprints of biological processes or past exposures to specific carcinogens. Mutational signatures can help to elucidate tumor evolution and have prognostic and therapeutic implications.
Mathematical estimation of signature activities is a high-dimensional statistical problem. Although many tools for signature analysis have been developed, a recent analysis showed that no tool performs best under all conditions.
We propose to identify the best-performing tool for a given task, cancer type, and number of mutations. This will be achieved by extensive prior testing of the existing tools on synthetic data under different conditions. We further propose to design a new tool that will explicitly include mutations that are not well explained by any known signature.
With the two proposed tools, we aim to optimally use costly sequencing data and produce informed decisions for patient treatments.
{kind=link}
Myeloid Malignancies
Meyer group Prof. Dr. med. Sara C. Meyer
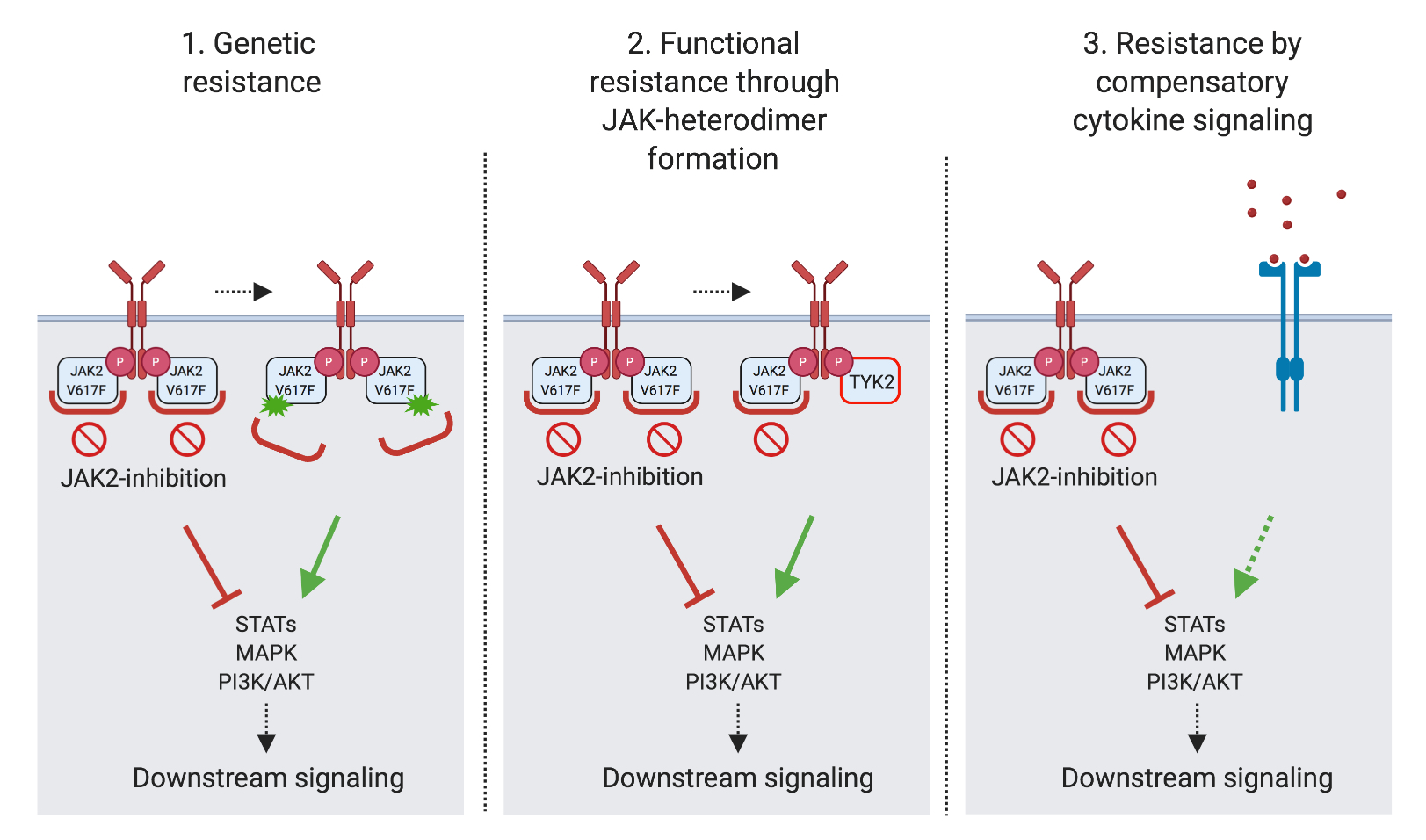
Myeloproliferative neoplasms (MPN) are chronic leukemias characterized by constitutive activation of JAK2 tyrosine kinase signaling. Clinical JAK2 inhibitors bring benefits for patients, but have limited disease-modifying activity. Allogeneic hematopoietic cell transplantation is the only curative treatment to date.
The Meyer lab has a specific interest in the oncogenic signaling driving MPN. We have demonstrated that activation of the MAPK pathway with MEK1/2 and ERK1/2 kinases, which is involved in several cancers, limits JAK2 inhibitor therapy and needs to be adressed to enhance efficacy (Stivala, JCI 2019; Brkic, Leukemia 2021). These findings have translated to a clinical study (Adore, NCT04097821).
Our lab is investigating mechanisms of resistance, which mediate loss of response to clinical JAK2 inhibitors, and approaches to overcome resistance. Notably, we are involved in the characterization of novel types of JAK2 inhibitors incl. type II JAK2 inhibitors currently in development towards clinical studies (Meyer, Cancer Cell 2015; Codilupi, CCR 2024).
{kind=link}
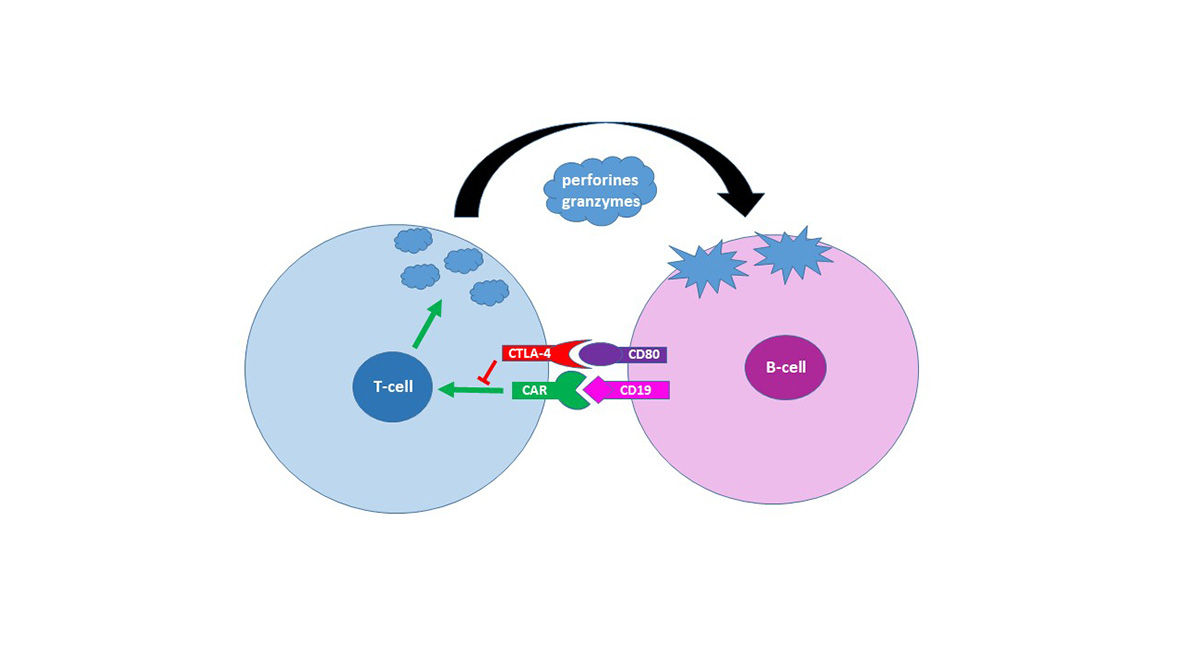
Genetic variants of immune checkpoint regulators as response biomarkers in B-cell lymphoma treated with CAR-T cell therapy
Pabst & Seipel group Prof. Dr. med. Thomas Pabst, PD Dr. Katja Seipel
Starting in 2020 CAR T-cell therapy has been applied in R/R B-cell lymphoma at Inselspital Bern. Biomarkers of response were evaluated in retrospective analysis in order to predict treatment outcome and improve CAR-T cell therapy.
{kind=link}