

Genomik
Forschungsprojekte
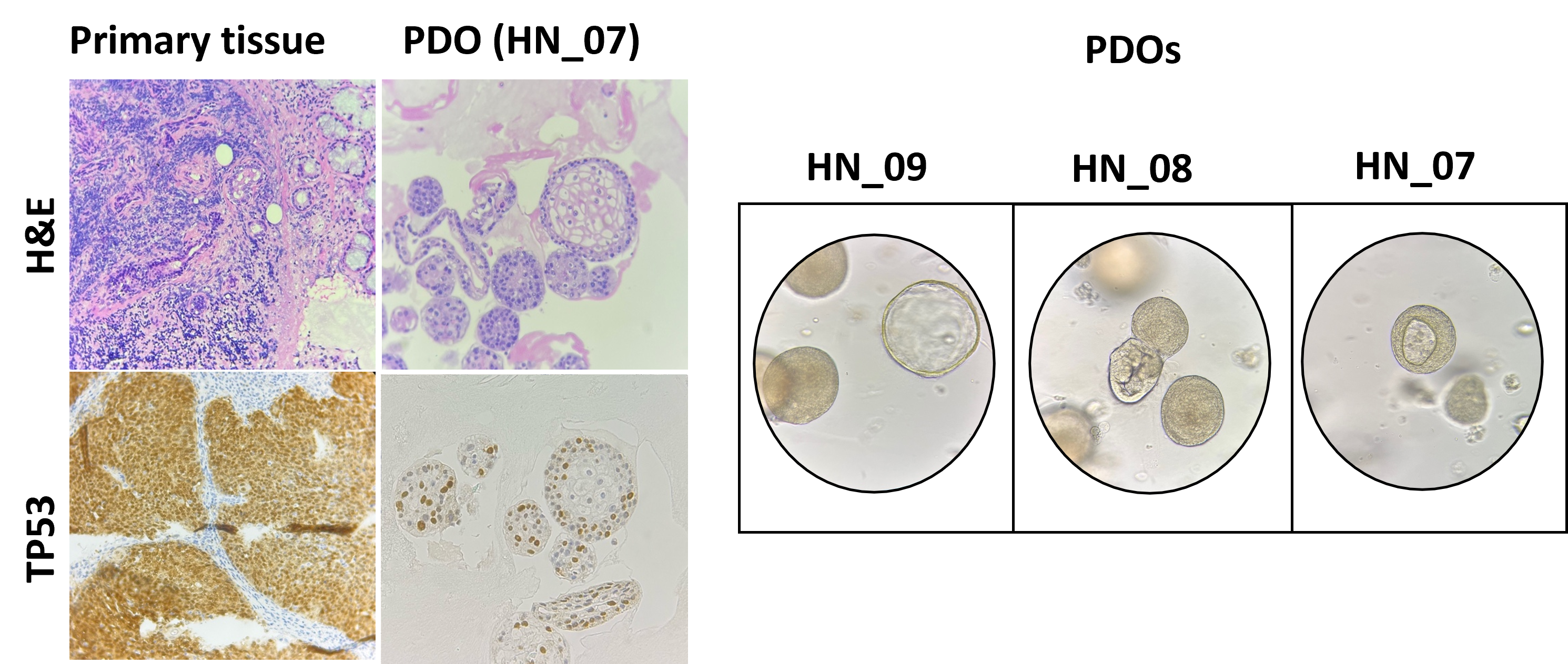
Eine personalisierte Multi-omics-Entdeckungs- und Validierungsplattform für rezidivierende Plattenepithelkarzinome im Kopf- und Halsbereich
{kind=link}
Gruppe Giger Head and Neck Anticancer Center, POLARES Research Group
Die diagnostischen und therapeutischen Entwicklungen der letzten Jahre haben die Prognose für Patienten mit Kopf-Hals-Plattenepithelkarzinomen (KH-PE-Karzinom) verbessert. Trotz dieser Entwicklungen erleidet ein erheblicher Anteil der Patienten, nach einem ersten Ansprechen auf die Standardbehandlung, einen Rückfall. Die Möglichkeiten einer Zweit-Behandlung sind beschränkt, und es fehlt an personalisierten Behandlungsansätzen, die die genomische/ epigenetische Landschaft des Tumors berücksichtigen.
Ziel dieser Studie ist es, ein Exzellenzzentrum für KH-PE-Karzinome einzurichten, das die Lücke zwischen genomischer Analyse und Umsetzung der Ergebnisse in klinische Studien schliesst. Durch die Etablierung einer Multi-omics-Entdeckungs- und Validierungsplattform, unter dem Dach des University Comprehensive Cancer Center Inselspital (UCI), will dieses Konsortium (HNO, Kopf- und Halschirurgie; Medizinische Onkologie; Radio-Onkologie) herausfinden, wie Veränderungen auf genomischer und epigenetischer Ebene die Karzinom-Entstehung, das Ansprechen auf die Behandlung und die Resistenz bei KH-PE-Karzinomen regulieren und dadurch neue Mechanismen zur Bekämpfung von Wiederauftreten identifizieren.
Im Namen des Konsortiums: Prof. Dr. Roland Giger (Lead), Otorhinolaryngology, Head and Neck Surgery; PD Dr. Olgun Eliçin, Radio-Oncology; Dr. Simon Häfliger, Medical Oncology; PD Dr. Michaela Medová, Radio-Oncology, DBMR; Prof. Dr. Carsten Riether, Medical Oncology, DBMR; Dr. Daniel H. Schanne, Radio-Oncology
Nicht-kodierende RNAs (ncRNAs) bei Lungenkrebs
{kind=link}
Gruppe Häfliger Dr. med. et phil. nat. Simon Häfliger
Lungenkrebs ist weltweit die häufigste krebsbedingte Todesursache. Unsere Forschung beschäftigt sich mit der faszinierenden Welt der nicht-kodierenden RNAs (ncRNAs), RNA-Moleküle, die nicht in Proteine übersetzt werden.
- Micro-RNAs (miRNAs): Diese winzigen RNA-Moleküle können entweder als Tumorsuppressoren oder als Onkogene wirken und die Krebsentwicklung beeinflussen.
- Long non-coding RNAs (lncRNAs): Diese längeren RNA-Moleküle beeinflussen die Genexpression und sind von zentraler Bedeutung für die epigenetische Regulierung.
Mittels öffentlicher Datenbanken, Blut von Lungenkrebspatienten, Krebszelllinien und bioinformatischen Analysen untersuchen wir ncRNAs mit der Fragestellung:
- Diagnostische Biomarker: Können zirkulierende ncRNAs als Frühindikatoren für Lungenkrebs dienen?
- Mechanismus: Wie beeinflussen diese ncRNAs das Fortschreiten von Krebs?
- Prädiktive und prognostische Marker: Können sie bei Behandlungsentscheidungen helfen?
Die Ergebnisse dieser Analysen versuchen wir in praktische klinische Anwendungen zu übersetzen, um Behandlungstrategien und damit das Überleben von Patientinnen und Patienten zu verbessern.
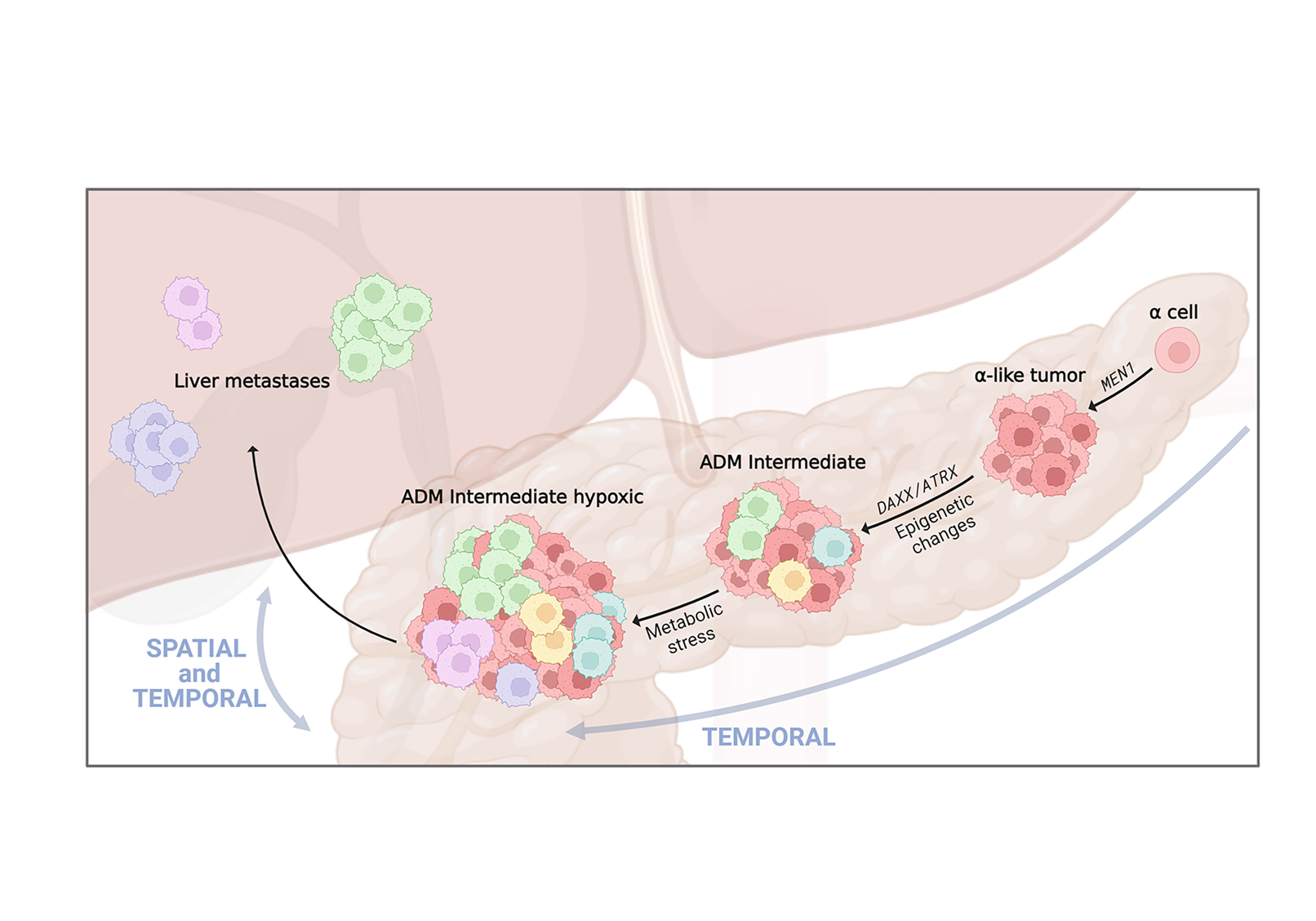
Entschlüsselung der Rolle der Heterogenität von Tumorzellen bei der Progression von neuroendokrinen Tumoren des Pankreas
Gruppe Marinoni Perren Sadowski PD Dr. Ilaria Marinoni, PhD, Prof. Dr. med. Aurel Perren, PD Dr. Martin Sadowski, PhD
Krebs ist eine dynamische Krankheit; genetische und epigenetische Veränderungen führen zu einer Heterogenität der Zellen innerhalb des Tumors, was zur Auswahl aggressiver Zellpopulationen führt, die das Fortschreiten der Krankheit und schliesslich die Metastasierung vorantreiben können.
Neuroendokrine Tumore der Bauchspeicheldrüse (PanNETs) sind Tumore, die von Zellen der Langerhans-Inseln stammen. Sie weisen eine intra-tumorale Zellheterogenität auf. Aber es ist unklar, wie diese während der Tumorentwicklung entsteht und sich entwickelt.
Unsere bisherigen Daten deuten darauf hin, dass epigenetische Veränderungen die Hauptursache für die Progression und Zellheterogenität bei PanNETs sind. Durch die Integration von epigenetischen und transkriptomischen Profilen konnten wir feststellen, dass die Dedifferenzierung von Zellen und metabolische Veränderungen das Fortschreiten von kleinen PanNETs zu fortgeschrittenen PanNETs charakterisieren.
Derzeit untersuchen wir die Entwicklung der intra-tumoralen Heterogenität von PanNETs über Raum und Zeit hinweg. Spezifische Zellsubpopulationen, die als treibende Kraft des Fortschreitens identifiziert wurden, könnten dann gezielt therapeutisch angegangen werden, um die Metastasenbildung zu stoppen.
{kind=link}
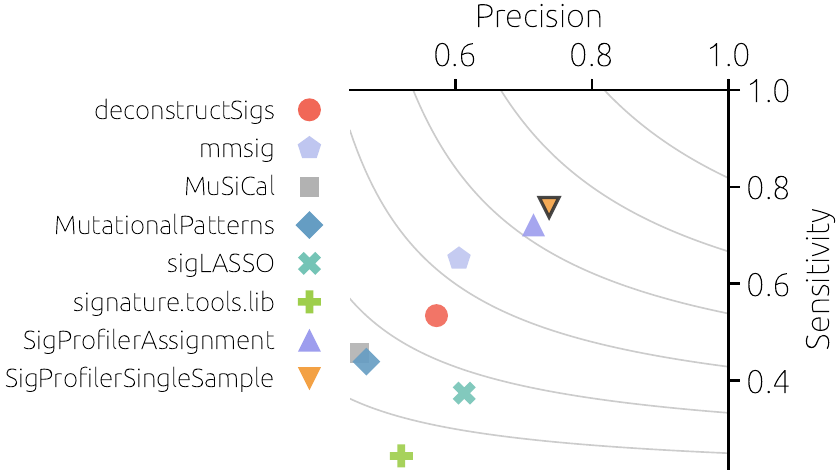
Einfache und genaue Quantifizierung von Mutationssignaturen in Tumoren
Gruppe Medo Prof. Dr. Matúš Medo
Mutationen im Genom sind oft nicht zufällig, sondern folgen bestimmten Mustern. Diese Muster, Mutationssignaturen genannt, sind Spuren biologischer Prozesse oder früherer Expositionen gegenüber bestimmten Karzinogenen. Mutationssignaturen können zur Aufklärung der Tumorentwicklung beitragen und prognostische und therapeutische Auswirkungen haben.
Die mathematische Schätzung von Signaturaktivitäten ist ein hochdimensionales statistisches Problem. Obwohl viele Tools zur Signaturanalyse entwickelt wurden, zeigte eine aktuelle Analyse, dass kein Tool unter allen Bedingungen die beste Leistung erbringt.
Wir schlagen vor, das leistungsstärkste Tool für eine bestimmte Aufgabe, Krebsart und Anzahl von Mutationen zu ermitteln. Dies wird durch umfangreiche vorherige Tests der vorhandenen Tools an synthetischen Daten unter verschiedenen Bedingungen erreicht. Wir schlagen ausserdem vor, ein neues Tool zu entwickeln, das Mutationen explizit einbezieht, die durch keine bekannte Signatur gut erklärt werden können.
Mit den beiden vorgeschlagenen Tools wollen wir kostspielige Sequenzierungsdaten optimal nutzen und fundierte Entscheidungen für die Behandlung von Patienten treffen.
{kind=link}
Myeloische Malignome
Gruppe Meyer Prof. Dr. med. Sara C. Meyer
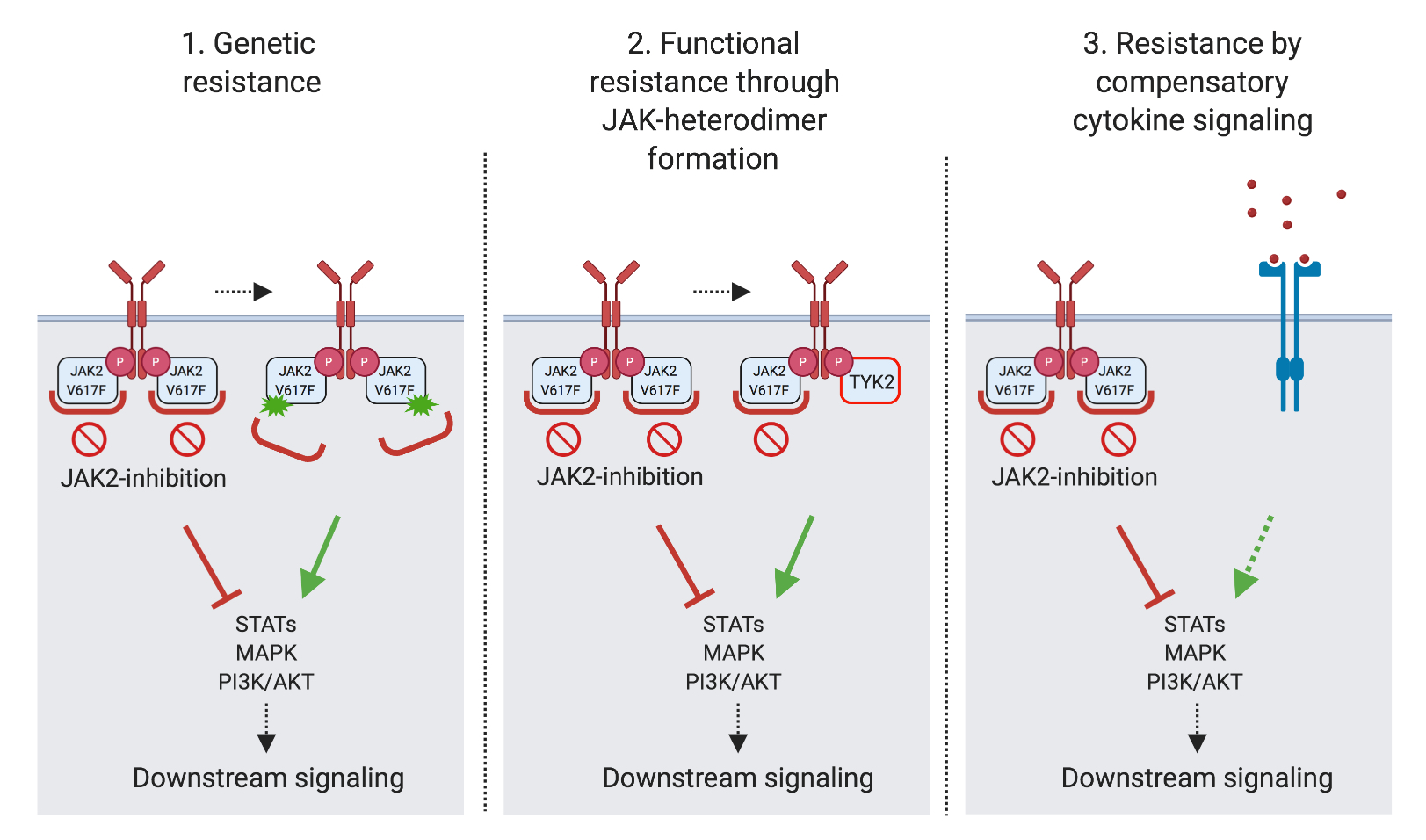
Myeloproliferative Neoplasien (MPN) sind chronische Leukämien. Sie sind durch eine konstitutive Aktivierung des JAK2 Tyrosinkinase-Signaling gekennzeichnet. JAK2 Inhibitoren stellen einen Standard in der Behandlung dar, haben aber nur einen begrenzte krankheitsmodizierenden Effekt. Die allogene hämatopoietische Stammzelltransplantation ist bisher der einzige Therapieansatz mit kurativem Potential.
Mein Forschungsteam untersucht das onkogene Signaling, das den MPN zugrunde liegt. Wir konnten zeigen, dass die Aktivierung des MAPK Signaling, eines Pathways, der bei verschiedenen Krebsarten eine wichtige Rolle spielt, bei MPN die Wirksamkeit der JAK2 Inhibitor-Behandlung vermindert und gezielt angegangen werden muss (Stivala, JCI 2019; Brkic Leukemia 2021).
Das hat Eingang in eine klinische Studie gefunden (Adore, NCT04097821). Weiter untersucht mein Forschungsteam Resistenzmechanismen, die zu Verlust des Therapieansprechens auf die JAK2 Inhibitoren führen. Insbesondere sind wir involviert in die Entwicklung neuer JAK2 Inhibitoren mit verbesserter Wirksamkeit bei MPN, die sich in Entwicklung zu klinischen Studien befinden (Meyer, Cancer Cell 2015; Codilupi, CCR 2024).
{kind=link}
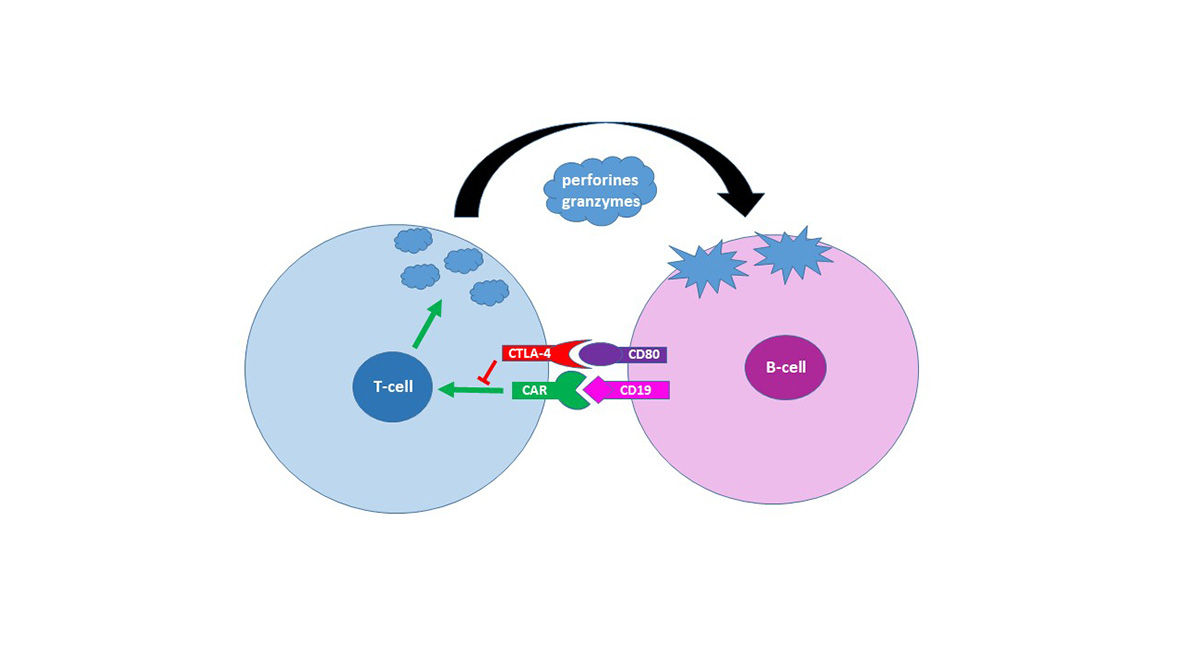
Genetische Varianten der Immunregulatoren als Response Biomarker bei B-Zell Lymphomen mit CAR-T Zell Therapie
Gruppe Pabst & Seipel Prof. Dr. med. Thomas Pabst, PD Dr. Katja Seipel
Seit 2020 werden am Inselspital Patienten mit B-Zell Lymphomen mit CAR-T Zell Therapie behandelt. Wir evaluieren das unterschiedliche Ansprechen auf CAR-T Zell Therapie und suchen Response Biomarker, die auf erfolgreiche Behandlung hinweisen können.
{kind=link}